An R function to removing qPCR outliers within technical replicates
The problem
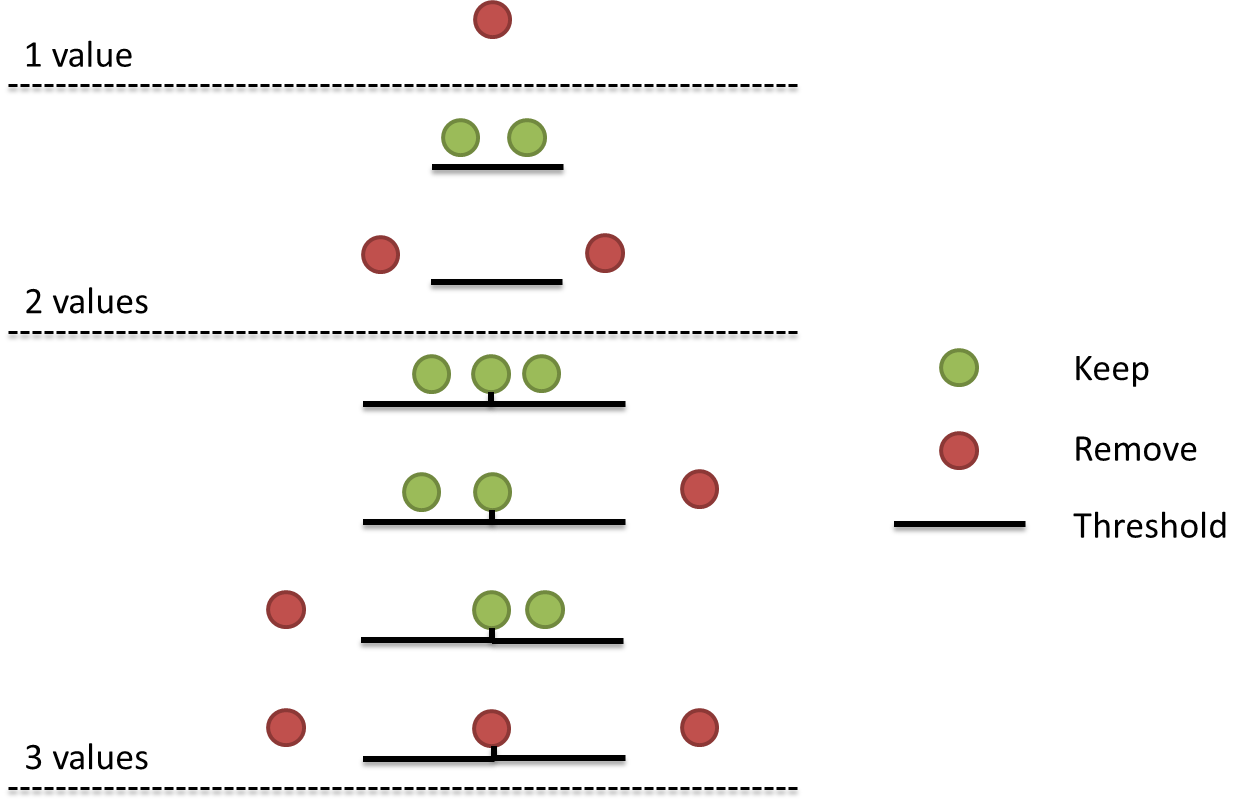
When performing qPCR you typically have three technical replicates of every sample. The Cq values within replicates are expected to be very close to each other. However, sometimes outliers occur thru for example, inaccurate pipetting, accidentally pipetting in the wrong well or seal detachment. Typically a Cq value more than 0.5 apart from the other two is considered an outlier. Some people prefer an ever stricter cutoff. On the other hand, if the target gene has very low expression - and thus high Cq values - high variation is unavoidable and a more lenient threshold might be preferred. In this post we will make a function that finds and removes outliers based on the deviation from the median value, given a certain threshold value. The function will use the following rules:
- If only one Cq value is present (i.e. the other replicates failed to produce a Cq value), it will be removed.
- If only two Cq values are present, they need to be less than a threshold apart.
- For three or more technical replicates:
- If the absolute distance between a Cq value and the median Cq is greater than a set threshold, than this value will be removed.
- If all Cq values within a technical replicate are more than a threshold apart, they will all be removed.
For manipulating the data we will mostly use functions from the tidyverse.
library(tidyverse)In this example we will use some randomly generated data. It contain Cq values for three genes: gene_a, gene_b and gene_hk (hk = housekeeping). There are three treatment conditions: a, b and ctrl, where ctrl is the untreated control. Three biological replicates are included. The data contains outliers representing all conditions previously specified, which we will try to remove.
The data is in a tidy format, meaning that every variable has its own column, and every measurement has its own row.
| treatment | bio_rep | tech_rep | primer_pair | cq_values |
|---|---|---|---|---|
| ctrl | 1 | 1 | gene_a | NA |
| ctrl | 1 | 2 | gene_a | 35.0 |
| ctrl | 1 | 3 | gene_a | NA |
| ctrl | 2 | 1 | gene_a | 34.9 |
| ctrl | 2 | 2 | gene_a | 34.3 |
First steps
For every sample we will calculate:
- The median Cq value.
- The number of replicates that have a value at all. The purpose of this step is to enable removing those samples where only one or no wells resulted in a Cq value.
median_cq <- raw_data %>%
# first group the data to perform the calculation for every sample separately.
group_by(treatment, primer_pair, bio_rep) %>%
# then calculate the median and count
summarise(median_cq = median(cq_values, na.rm = TRUE),
count = sum(!is.na(cq_values))) | treatment | primer_pair | bio_rep | median_cq | count |
|---|---|---|---|---|
| a | gene_a | 1 | 22.3 | 3 |
| a | gene_a | 2 | 21.7 | 3 |
| a | gene_a | 3 | 21.5 | 3 |
| a | gene_b | 1 | 22.1 | 3 |
| a | gene_b | 2 | 22.3 | 3 |
Next we will join the table back to the original data, so that the median and count will become their own column. We can then simply remove those samples with only one or zero technical replicates, and calculate the distance from the median for every remaining technical replicate. Finally, a column is created that contains TRUE if the distance from the median is within a threshold value or FALSE if it is not.
Note: if there are only two technical replicates within a sample, the median falls exactly between these two values. We want the two replicates to be less than the threshold apart. To get the distance between the replicates we multiply the distance from the median by 2.
#As an example we will use a threshold value of 1.
#This is probably too high for most practical applications.
threshold <- 1
dev_test <- raw_data %>%
full_join(median_cq, by = c("treatment", "primer_pair", "bio_rep")) %>%
# take out samples with only 1 or 0 tech reps
filter(count > 1) %>%
# calculate the distance from the median
mutate(distance_med = abs(cq_values - median_cq)) %>%
# check if the distance from the median is within the threshold.
mutate(keep = if_else(count == 2,
if_else(distance_med*2 < threshold, TRUE, FALSE),
if_else(distance_med < threshold, TRUE, FALSE)))| treatment | bio_rep | tech_rep | primer_pair | cq_values | median_cq | count | distance_med | keep |
|---|---|---|---|---|---|---|---|---|
| ctrl | 2 | 1 | gene_a | 34.9 | 34.5 | 3 | 0.4 | TRUE |
| ctrl | 2 | 2 | gene_a | 34.3 | 34.5 | 3 | 0.2 | TRUE |
| ctrl | 2 | 3 | gene_a | 34.5 | 34.5 | 3 | 0.0 | TRUE |
| ctrl | 3 | 1 | gene_a | 34.3 | 34.3 | 3 | 0.0 | TRUE |
| ctrl | 3 | 2 | gene_a | 34.7 | 34.3 | 3 | 0.4 | TRUE |
There is one condition left we need to be able to remove, namely where all three technical replicates are more than the threshold apart from each other. Using the table we have generated so far the middle replicate will be kept, because it is the median and therefore the distance from the median is 0. To get rid of this we will only keep samples where at least two replicates are marked as TRUE in the keep column.
# count the number of TRUEs
count_true <- dev_test %>%
group_by(treatment, primer_pair, bio_rep) %>%
summarise(count_keep = sum(keep, na.rm = TRUE)) %>%
ungroup()
# finally, filter out all outliers
clean_data <- dev_test %>%
full_join(count_true, by = c("treatment", "primer_pair", "bio_rep")) %>%
# remove outliers
filter(keep == TRUE,
count_keep > 1) %>%
# remove the now unnecessary columns
select(-(median_cq:count_keep)) | treatment | bio_rep | tech_rep | primer_pair | cq_values |
|---|---|---|---|---|
| ctrl | 2 | 1 | gene_a | 34.9 |
| ctrl | 2 | 2 | gene_a | 34.3 |
| ctrl | 2 | 3 | gene_a | 34.5 |
| ctrl | 3 | 1 | gene_a | 34.3 |
| ctrl | 3 | 2 | gene_a | 34.7 |
To find all Cq values that have been removed:
outliers <- raw_data %>%
setdiff(clean_data)| treatment | bio_rep | tech_rep | primer_pair | cq_values |
|---|---|---|---|---|
| ctrl | 1 | 1 | gene_a | NA |
| ctrl | 1 | 2 | gene_a | 35.0 |
| ctrl | 1 | 3 | gene_a | NA |
| a | 1 | 2 | gene_a | 25.0 |
| b | 1 | 3 | gene_a | 20.0 |
| ctrl | 1 | 2 | gene_b | NA |
| a | 2 | 1 | gene_b | 18.0 |
| a | 2 | 2 | gene_b | 22.3 |
| a | 2 | 3 | gene_b | 27.0 |
| b | 1 | 1 | gene_b | 26.0 |
| b | 1 | 2 | gene_b | 24.0 |
| b | 1 | 3 | gene_b | NA |
Turning it into a function
Throughout this post we have almost exclusively use functions from the tidyverse to manipulate the data. Using tidyverse function inside custom functions is possible, but we do need the rlang package.
library(rlang)Our function will have the following arguments:
- The first argument is name of the data we want to use.
cq =tells the function the name of the column containing the Cq values.- With
threshold =we can set the maximum distance from the median. - Finally, the function needs the names of all other columns that are not the cq_values or denote technical replicates. These will be used to make groups. Calculations will be made for each unique combination.
Which and how many experimental variables there are depends on the experimental setup. Therefore, the final argument of the function is .... Now we can supply any number of column names (unquoted and separated by a comma). Inside the qpcr_clean() function, ... can be passed to group_by(). There is just one problem. The tidyverse function full_join() needs the same set of column names to join the tables by, but as a character vector. Luckely ... can be converted to such a vector with:
dots <- names(enquos(..., .named = TRUE))Finally the name of the column containing the Cq values can be passed to the tidyverse functions by surrounding cq with double braces: {{cq}}.
qpcr_clean <- function(.data, cq, threshold, ...){
# to do: add if statements to check input data etc.
# convert ... to character vector
dots <- names(enquos(..., .named = TRUE))
# calculate the median and count for every sample
median_cq <- .data %>%
group_by(...) %>%
summarise(median_cq = median({{cq}}, na.rm = TRUE),
count = sum(!is.na({{cq}}))) %>%
ungroup()
# remove solo tech reps, test if distance from the median is < threshold
dev_test <- .data %>%
full_join(median_cq, by = dots) %>%
filter(count > 1) %>%
mutate(distance_med = abs({{cq}} - median_cq)) %>%
mutate(keep = if_else(count == 2,
if_else(distance_med*2 < threshold, TRUE, FALSE),
if_else(distance_med < threshold, TRUE, FALSE)))
# count the # of TRUEs per sample
count_true <- dev_test %>%
group_by(...) %>%
summarise(count_keep = sum(keep, na.rm = TRUE)) %>%
ungroup()
# remove all outliers
clean_data <- dev_test %>%
full_join(count_true, by = dots) %>%
filter(count_keep > 1,
keep == TRUE) %>%
select(-(median_cq:count_keep))
clean_data
}Lets test if it works:
clean_data <- qpcr_clean(raw_data,
cq = cq_values,
threshold = 1,
treatment, primer_pair, bio_rep)| treatment | bio_rep | tech_rep | primer_pair | cq_values |
|---|---|---|---|---|
| ctrl | 2 | 1 | gene_a | 34.9 |
| ctrl | 2 | 2 | gene_a | 34.3 |
| ctrl | 2 | 3 | gene_a | 34.5 |
| ctrl | 3 | 1 | gene_a | 34.3 |
| ctrl | 3 | 2 | gene_a | 34.7 |
Outlier context
For context, you might want to see the other Cq values within a technical replicate that contains an outlier. For this we can make a separate function called qpcr_outlier_context().
The function arguments will be:
- With
raw_data =we supply the raw unfiltered data - With
clean_data =we supply the cleaned data from theqpcr_clean()function cq_values =is used to give name of the column containing the cq valuestech_rep =gives the name of the column containing the technical replicate information...all other column names, excluding the columns containing the Cq values or technical replicates.
qpcr_outlier_context <- function(raw_data, clean_data, cq_values, tech_rep, ...){
# turn some values into strings for joining tables
dots <- names(enquos(..., .named = TRUE))
string_tech_rep <- names(enquos(tech_rep, .named = TRUE))
join2 <- append(dots, string_tech_rep)
# first find all the outliers
outliers <- raw_data %>%
setdiff(clean_data)
# give outliers extra columns, all TRUE
extra_col <- outliers %>%
mutate(outlier = TRUE) %>%
select(-{{cq_values}})
# find all triplets with outliers
triplets <- outliers %>%
# remove some columns to prevent double columns after joining
select(-{{cq_values}}) %>%
select(-{{tech_rep}}) %>%
# left_join() every sample not in outliers
left_join(raw_data, by = dots) %>%
distinct() %>%
# add a column called `outlier` that marks outliers with TRUE
left_join(extra_col, by = join2)
triplets
}Lets try it out!
outlier_triplets <- qpcr_outlier_context(raw_data = raw_data,
clean_data = clean_data,
cq_values = cq_values,
tech_rep = tech_rep,
treatment, bio_rep, primer_pair)| treatment | bio_rep | primer_pair | tech_rep | cq_values | outlier |
|---|---|---|---|---|---|
| ctrl | 1 | gene_a | 1 | TRUE | |
| ctrl | 1 | gene_a | 2 | 35.0 | TRUE |
| ctrl | 1 | gene_a | 3 | TRUE | |
| a | 1 | gene_a | 1 | 21.7 | |
| a | 1 | gene_a | 2 | 25.0 | TRUE |
| a | 1 | gene_a | 3 | 22.3 | |
| b | 1 | gene_a | 1 | 25.3 | |
| b | 1 | gene_a | 2 | 25.5 | |
| b | 1 | gene_a | 3 | 20.0 | TRUE |
| ctrl | 1 | gene_b | 1 | 35.1 | |
| ctrl | 1 | gene_b | 2 | TRUE | |
| ctrl | 1 | gene_b | 3 | 35.6 | |
| a | 2 | gene_b | 1 | 18.0 | TRUE |
| a | 2 | gene_b | 2 | 22.3 | TRUE |
| a | 2 | gene_b | 3 | 27.0 | TRUE |
| b | 1 | gene_b | 1 | 26.0 | TRUE |
| b | 1 | gene_b | 2 | 24.0 | TRUE |
| b | 1 | gene_b | 3 | TRUE |
We indeed find back every kind of outlier condition specified in the beginning of this post. Now we don’t have to visually inspect our qPCR results in excel anymore!